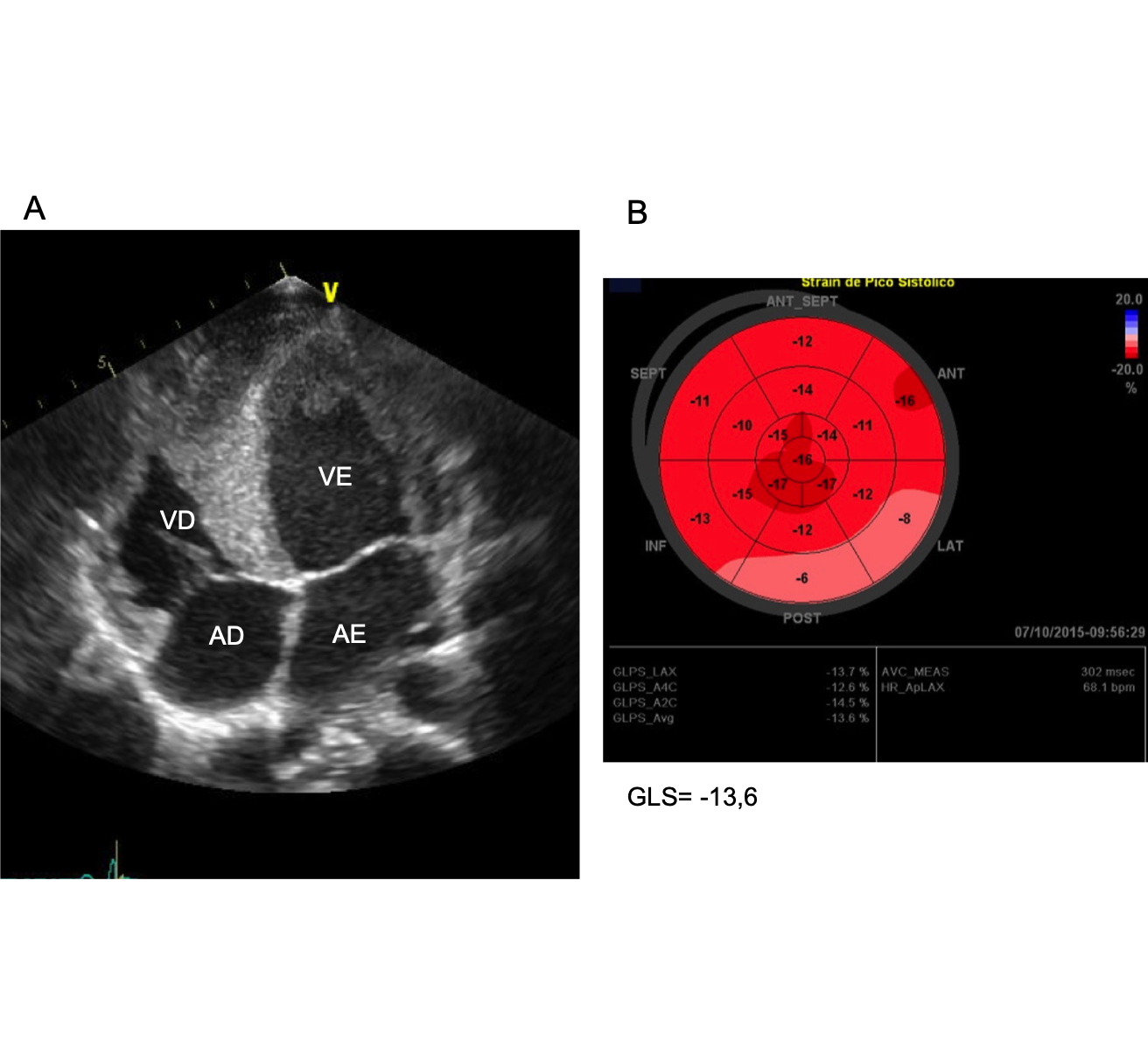
R.A.G.E, 21 anos, masculino manifestou precordialgia típica e dispneia aos moderados esforços. Havia ainda história de morte súbita na família. Ao exame físico, notava-se um fenótipo leptossômico, ictus desviado a esquerda com impulsão precordial visível, turgência jugular a 90 graus e edema de MMII. O ecocardiograma transorácico (ECOTT) inicial revelou hipertrofia concêntrica do VE de grau moderado, com septo de 14mm e de aspecto hiperrefringente; um discreto déficit sistólico do VE, com fração de ejeção (FEVE) de 44% e redução no "strain" (GLS=-13,6%) (Figura 1). A ressonância magnética cardíaca (RMC) confirmou os achados da ECOTT e mostrou padrão de fibrose miocárdica sugestivo de doença infiltrava. Não havia lesões obstrutivas coronárias (CATE). A biópsia miocárdica revelou fibrose subendocárdica associada a alterações sarcoplasmáticas. A pesquisa para proteína amiloide foi negativa, assim como para doença de Fabry. Paciente foi então acompanhado como provável cardiomioatia (CMP) hipertrófica sarcomérica embora teste genético não tenha sido realizado. Ao longo de 4 anos, evoluiu com perda de peso progressiva e piora da classe funcional. Novo ECOTT evidenciou piora da FEVE (27%). Clinicamente foi detectado déficit auditivo bilateral e perda de força muscular periférica. A biópsia de músculo esquelético demonstrou alterações típicas de mitocondriopatia e teste genético confirmou mutação tipo 3242A>G . Paciente recebeu tratamento otimizado para ICFER e acompanhamento multidisciplinar com Neurologia, mantendo-se estável clinicamente.
DISCUSSÃO: As CMP hipertróficas são um grupo heterogêneo de doenças que se caracteriza pela presença de aumento da espessura miocárdica e frequentemente disfunção ventricular diastólica mas não sistólica. A etiologia, embora seja sarcomérica em cerca de 40% dos casos, pode ser secundária diversas outras doenças sistêmicas. Faz-se necessária a atenção do cardiologista para outras etiologias, especialmente em cenário clínico de doenças sistêmicas e na evolução para disfunção sistólica de VE, como no caso apresentado. Na suspeita de mitocondriopatias a investigação diagnóstica deve compreender exames de imagem cardiovascular de multimodalidades, testes genéticos, biópsia de músculo periférico e a biópsia miocárdica pode ser necessária. Em pacientes com mitocondriopatias, a prevalência do envolvimento cardíaco chega a 30% e a taxa de eventos adversos cardiovasculares é estimada em até 10% em 7 anos, sendo a presença de hipetrofia miocárdica determinante prognóstico.